
Immunohistochemical detection and localisation of prion protein in brain tissue of cattle, sheep and goats
I.Kolačko, S. Zrnčić, Š. Naletilić*, B. Iulini and K. Branović Čakanić
Ivana KOLAČKO1, kolacko@veinst.hr, orcid.org/0009-0003-2209-1126; Snježana ZRNČIĆ2, zrncic@veinst.hr, orcid.org/0000-0003-4084-5641; Šimun NALETILIĆ3* (corresponding author), naletilic@veinst.hr, orcid.org/0009-0002-5805-9892; Barbara IULINI 4, barbara.iulini@izsplv.it; Karmen BRANOVIĆ ČAKANIĆ1, branovic@veinst.hr, orcid.org/0000-0001-8760-4680.
1Laboratory for Transmissible Spongiform Encephalopathies, Department of Pathological Morphology, Croatian Veterinary Institute, 10000 Zagreb, Croatia
2Laboratory for Aquatic Animal Diseases, Department of Pathological Morphology, Croatian Veterinary Institute, 10000 Zagreb, Croatia
3Laboratory for Pathology, Department of Pathological Morphology, Croatian Veterinary Institute, 10000 Zagreb, Croatia
4Veterinary Medical Research Institute for Piedmont, Liguria and the Aosta Valley, Laboratory of Neuropathology, 10154 Turin, Italy
![]()
https://doi.org/10.46419/cvj.57.3.6
Abstract
The misfolding of the cellular prion protein into its pathogenic isoform prion—a proteinaceous infectious particle-causes fatal neurodegenerative diseases. These prion diseases are classified as transmissible spongiform encephalopathies, which may arise from genetic mutations, infectious transmission or sporadic occurrence. They are characterised by protein aggregation and progressive neurodegeneration. In humans, prion diseases include Kuru, Creutzfeldt-Jakob disease, variant Creutzfeldt-Jakob disease, Gerstmann-Sträussler-Scheinker-Syndrome, and fatal familial insomnia. In animals, they include atypical and classical scrapie in sheep and goats, atypical and classical bovine spongiform encephalopathy, also known as “mad cow disease”, and chronic wasting disease in cervids. Although the mechanisms underlying neurodegeneration and the role of neuroinflammation in these disorders remain insufficiently understood, characteristic features include spongiform changes, neuronal loss, gliosis, and accumulation of the pathogenic isoform of the prion. To date, there is no reliable ante mortem diagnostic test for these encephalopathies. The diagnosis is confirmed postmortem, and immunohistochemistry is one of the key diagnostic techniques. This review article summarises the available research data about prion diseases, focusing on the detection and localisation of the pathogenic isoform of the prion protein in brain tissue of cattle, sheep, and goats using immunohistochemical analysis.
Key words: prion; transmissible spongiform encephalopathy; scrapie; bovine spongiform encephalopathy; immunohistochemistry
Introduction
A prion is defined as a “proteinaceous infectious particle,” responsible for the transmission of a group of fatal neurodegenerative diseases that affect humans and many other mammals (Prusiner, 1982). Disorders known as transmissible spongiform encephalopathies (TSEs) are characterised by the accumulation of an abnormal, disease-associated isoform of the host-encoded prion protein (PrPSc) in infected tissues.
Unlike the normal cellular form of the prion protein (PrPC), the isoform PrPSc is partially resistant to protease digestion and is associated with neuropathological changes characteristic of prion diseases (Chesebro, 1999). Disease-specific accumulation of PrPSc in affected brain tissue is detected using immunohistochemistry (IHC) on formalin-fixed samples. Accurate diagnosis relies on the recognition of distinct, disease-specific immunolabeling patterns, their cellular associations and neuroanatomical distribution. These features support confirmatory diagnosis in classical (Ryder et al., 2001) and atypical (Benestad et al., 2008) forms of scrapie and bovine spongiform encephalopathy (BSE). The roles and responsibilities of national reference laboratories (NRLs) and the European Union Reference Laboratory (EURL) for transmissible spongiform encephalopathies (TSEs) are outlined in Annex X of the TSE Regulation (https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32001R0999). The EURL (https://www.eurl-tse.eu/) is responsible for coordinating regular interlaboratory proficiency testing to evaluate the performance of NRLs across the European Union (EU). The list of antibodies and the protocol that is proven to be of use for IHC, can be found at the UK WOAH Reference Laboratory website (https://science.vla.gov.uk/tseglobalnet/confirmatory-diagnosis.html).
Classical BSE
Multiple morphological patterns of PrPSc deposition have been identified in the brains of cattle affected by classical BSE:
Glial type: Characterised by PrPSc deposits radiating from the nuclei of glial cells along their processes, conferring them a stellate appearance. This pattern is predominantly observed in central grey matter and cerebral lamina, and also within medial pontine nuclei of the cerebral cortex, thalamus, and obex (Casalone et al., 2006).
Granular type: Marked by numerous small PrPSc deposits predominantly located within the neuropil of grey matter nuclei, including the dorsal motor nucleus of the vagus (DMV), the nucleus of the solitary tract (NST), and various thalamic nuclei (Casalone et al., 2006).
Intraneuronal type: PrPSc deposits are frequently found throughout the neuronal cytoplasm, notably within the DMV, reticular formation, olivary nuclei, vestibular complex, pontine nuclei, thalamic nuclei, and the hypothalamus (Casalone et al., 2006).
Perineuronal type: Characterised by PrPSc deposits encircling individual neuronal perikarya and neurites, particularly within the caudate and putamen nuclei of the basal ganglia and the DMV. In addition, linear PrPSc deposition along neuronal processes is prominently observed, especially in the reticular formation of the brainstem (Casalone et al., 2006).
Coalescing type: In this pattern, large granular PrPSc deposits merge to form amorphous or mesh-like aggregations (Wilham et al., 2010).
Intraglial type: Consists of fine, punctate PrPSc deposits adjacent to glial cell nuclei. Notably, a hallmark of natural BSE cases is the preferential accumulation of PrPSc in the more rostral brain regions, in contrast to the distribution pattern typically seen in classical BSE (Lombardi et al., 2008).
Atypical BSE
Atypical BSE manifests in two distinct forms which include L-type (L-BSE) and H-type (H-BSE) prions. Atypical BSE prions were first identified in 2004 in France and Italy and subsequently classified based on the lower or higher apparent molecular weights of unglycosylated, protease-resistant form of the pathogenic prion protein (PrPSc) as detected by Western blot analysis (Biacabe et al., 2004).
The L-BSE variant, also known as bovine amyloidotic spongiform encephalopathy (BASE), is characterised by the atypical deposition of amyloid plaques within the brain (Casalone et al., 2004). In both H-BSE and L-BSE, PrPSc has been detected in central nervous system (CNS) tissue, peripheral ganglia and nerves, muscles (muscle spindles), adrenal glands, and the retina. Experimental transmission studies have shown that lymphoid tissue or gastrointestinal tissue consistently tested negative in atypical cases (EFSA, 2014a). Furthermore, findings from an interspecies transmission study involving L-BSE indicated that prion propagation occurs within the CNS, with centrifugal spread along peripheral nerve pathways (Iwamaru et al., 2010). At the level of the obex, the quantity and distribution of PrPSc immunolabelling are broadly comparable between atypical and classical BSE cases. However, subtle phenotype-specific differences can be identified. In H-BSE, PrPSc immunolabelling is predominantly localised to white matter tracts, whereas in L-BSE, smaller and more diffusely distributed deposits of immunolabelled PrPSc are observed within the reticular formation (Konold et al., 2012). Throughout other regions of the neuroaxis, both H-BSE and L-BSE show widespread PrPSc immunolabelling, although the distribution patterns observed differ distinctly from those characteristic for classical BSE (Stack et al, 2011). The cerebellum represents a key neuroanatomical target site for the distinguishing between atypical forms of BSE (Konold et al, 2012).
L-BSE is characterised by extensive deposition of small, plaque-like PrPSc accumulation, representing a distinctive feature of this atypical form. These deposits are predominantly localised within the thalamus, subcortical white matter, deeper cortical layers of the cerebrum, and the olfactory bulb (Casalone et al., 2004). The amyloid plaques associated with PrPSc in L-BSE are typically dense, predominantly unicentric, though multicentric forms may also occur. Morphologically, they appear as round structures up to 25 µm in diameter, characterised by a pale central core surrounded by a darker radially oriented periphery. Beyond these amyloid plaques, other patterns of PrPSc accumulation have been described in BASE cases. These include granular deposits, which are mildly present in the hypoglossal and olivary nuclei, and more prominently observed in the DMV, NST, its ventral subdivision (NSTV), and the reticular formation. Moreover, various morphological variants of PrPSc such as stellate, punctate, glial, intraneuronal, perineuronal, and linear deposits have been observed across multiple brain regions (Corona et al., 2017).
In L-BSE cases, PrPSc immunolabelling is also evident in both the molecular and granular layers of the cerebellum, closely resembling the deposition pattern reported in atypical scrapie in sheep (Orge et al., 2015).
In contrast, experimental models of H-BSE demonstrate minimal and less uniformly distributed PrPSc immunolabelling within the cerebellar layers. However, notable and widespread glial labelling is observed throughout the white matter of both the spinal cord and cerebellum (Konold et al., 2012). In naturally occurring H-BSE, the most characteristic PrPSc deposition patterns within the brainstem include fine granular, intraneuronal, linear, intraglial, and punctate forms. These deposits are predominantly localised at the level of the DMV, NST and its ventral part (NSTV), as well as the reticular formation, although some variability in their distribution has been noted (Porcario et al., 2011).
Classical scrapie
The medulla oblongata at the level of the obex is recognised as the earliest and most consistently affected neuroanatomical region exhibiting vacuolar changes in classical scrapie (CS). It represents the most reliable diagnostic site within the central nervous system using either histopathological assessment of vacuolation or immunohistochemical detection of PrPSc (Wood et al., 1997). In atypical scrapie, the medulla is only minimally affected, while the cerebellum, thalamus and basal ganglia are more pervasive and clearly affected (Benestad et al., 2008; Moore et al., 2008). Based on several studies, distinct types of PrPSc have been defined (Moore et al., 2008):
Intracellular:
Intraneuronal: PrPSc deposits, varying from fine granular to coarse and occasionally confluent forms, are localised within the cytoplasm of neuronal perikarya, typically surrounding the nucleus.
Intraglial: Dense granular or ovoid PrPSc accumulations, typically larger than neuronal deposits, are located adjacent to glial cell nuclei.
Extracellular:
Stellate (glial type): Radiating, branching PrPSc deposits centred around a prominent glial nucleus, with characteristic star shaped morphology (Figure 1).
Perivascular: PrPSc deposits localised circumferentially around blood vessels within the white matter.
Subpial: Loosely arranged to amorphous, multilocular continuous accumulations of PrPSc situated beneath the pia mater.
Subependymal: Resembling the perivascular pattern, this PrPSc deposition occurs within the glial layer beneath the ependymal lining of the ventricular system, typically appearing as discontinuous accumulations, predominantly localised around the lateral ventricles.
Linear: PrPSc deposits distributed in linear feature along the neuropil, often following neuronal processes.
Fine granular (fine punctate or fine particulate): Numerous small PrPSc deposits diffusely dispersed throughout the neuropil, creating a finely speckled appearance.
Aggregates (coarse granular, coarse particulate): Large, amorphous clusters of PrPSc irregularly distributed within the neuropil.
Perineuronal: PrPSc accumulations encircling individual, scattered neuronal perikarya and neurites.
Plaque-Like (plaques, vascular plaques): Relatively large, fibrillar PrPSc deposits with a radiating pattern, frequently localised around blood vessels of various diameters.
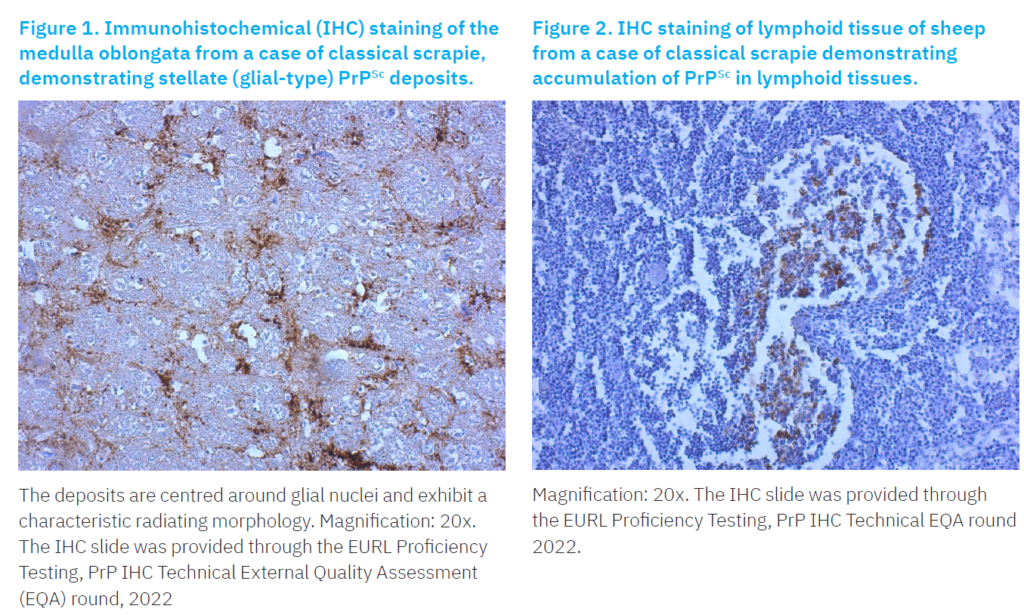
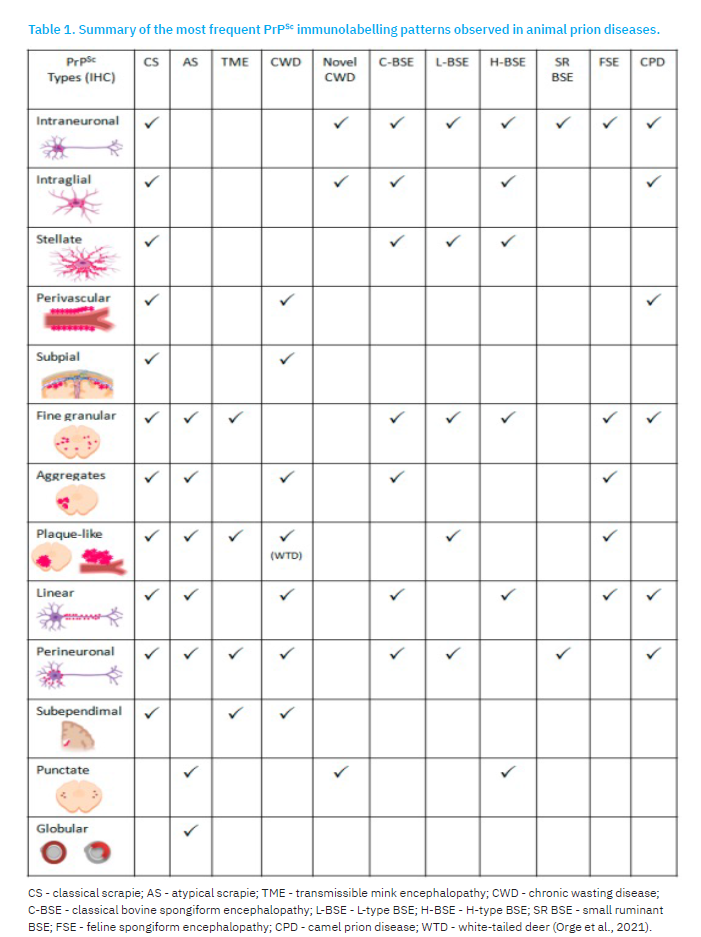
Although not consistently observed in all CS affected sheep, PrPSc deposits have also been detected in lymphoid tissues (Figure 2), including the tonsils, spleen, and lymph nodes, and other organs such as skeletal muscle, placenta, skin, mammary glands, distal ileum, proximal colon, pancreas, heart, and urinary bladder (EFSA, 2014b). PrPSc immunolabeling observed in CS-affected goats resembled those described for scrapie in sheep. However, in goats, the distribution of PrPSc appears to be more extensive and variable (González et al., 2009). This variability may be influenced by factors such as animal’s PRNP genotype, age, clinical stage, and even the specific prion strain involved (Sofianidis et al., 2006).
Atypical scrapie
The distribution of PrPSc in atypical scrapie (AS), as revealed by immunohistochemistry, differs significantly from that observed in CS. In AS, PrPSc deposition in the obex is typically mild, and primarily localised to the neuropil within the nucleus of the spinal tract of the trigeminal nerve and adjacent white matter tracts. In contrast, more intense and widespread PrPSc accumulation is commonly observed in the cerebellum, substantia nigra, thalamus, and basal nuclei, often occurring with or without associated vacuolation (Benestad et al., 2003). Within the cerebellum, PrPSc immunoreactivity may be evident in both the molecular and granular layers (Figure 3), or localised to only one of these regions (Orge et al., 2010).

A limited number of PrPSc morphological types have been identified in AS, including fine granular, aggregate, plaque-like, linear, perineuronal, and distinctive white matter-associated forms such as punctate and globular deposits. Among these, the fine granular and aggregate patterns are the most frequently observed, particularly in the rostral midbrain and thalamus, and are thought to represent PrPSc associated with synaptic regions or distal neuronal processes (Moore et al., 2008).
Intraneuronal and glial associated PrPSc types are notably absent in AS, likely due to the higher sensitivity of PrPSc to proteinase-K digestion compared to that in CS. This suggests a greater cellular capacity to digest the abnormal prion protein in atypical cases (Greenlee et al., 2019). In the white matter, two predominant deposition types are described: 1) the globular type, characterised by complete or crescent shaped, ring or oval-shaped accumulations, sometimes with irregular outline, and 2) the punctate type, consisting of small, discrete circular or tear-drop shaped deposits, also displaying irregular outlines (Moore et al., 2008).
Although conventional diagnostic methods typically fail to detect PrPSc in the peripheral or lymphoid tissues of sheep affected with AS, infectivity has been detected in the ileum, spleen, skeletal muscle, lymphoid tissues, and peripheral nerves (Andréoletti et al., 2011). Bioassays using transgenic mice have confirmed the presence of infectivity in lymphoid tissues, neural structures, and muscular tissue (Benestad et al., 2008). AS has also been reported in goats, exhibiting a similar electrophoretic PrPSc profile comparable to that observed in AS-affected sheep. However, the distribution of PrPSc in goats tends to be more rostrally located and involves anterior brain regions (Seuberlich et al., 2007).
Conclusion
At the level of the obex, no significant differences have been observed in the quantity or distribution of PrPSc immunostaining between atypical BSE and classical BSE. However, Konold et al. (2012) were the only researchers to demonstrate subtle phenotype-specific variations in this region. In contrast, the cerebellum has been identified as a more reliable tissue for differentiating between BSE subtypes. L-BSE is characterised by a diffuse and homogenous PrPSc immunostaining pattern within both the molecular and granular layers of the cerebellum, with prominent perineuronal deposits in the granular layer. H-BSE is characterised by strong PrPSc immunolabelling typically localised within the white matter of the spinal cord. Classical BSE, on the other hand, demonstrates more variable immunoreactivity patterns that contribute to its differentiation. In CS, the medulla oblongata at the level of the obex represents the most consistent and diagnostically relevant CNS region. In AS, PrPSc accumulation is more consistently and prominently observed in the cerebellum, thalamus, and basal ganglia, distinguishing it from classical forms.
IHC examination of brain tissue remains one of the gold standard techniques for the post-mortem confirmation of TSE diagnosis. IHC is considered a highly sensitive and specific method for the detection of PrPSc and is currently the most commonly used confirmation method in both passive and active surveillance programmes. Its diagnostic value is especially notable in cases lacking significant vacuolisation, as it enables the detection of PrPSc even in the absence of overt histopathological lesions. Nevertheless, while more reliable than conventional histology, IHC interpretation can be subjective, and variability in the assessment of PrPSc staining patterns may influence diagnostic outcomes leading to inconsistencies in reported results.
Acknowledgments
We extend our sincere thanks to Giuseppe Ru and the entire EURL team (Centro di Referenza Nazionale per le Encefalopatie Animali, Istituto Zooprofilattico Sperimentale di Torino) for providing the monitoring data. We also gratefully acknowledge our technician, Mihaela Stuparić Komušar, for her support in preparing the material used in the EURL PrP IHC Tehnical EQA round 2022 proficiency test.
References [… show]
Imunohistokemijska identifikacija i distribucija prion proteina u moždanom tkivu goveda, ovaca i koza
Ivana KOLAČKO 1, kolacko@veinst.hr, orcid.org/0009-0003-2209-1126; Snježana ZRNČIĆ 2, zrncic@veinst.hr, orcid.org/0000-0003-4084-5641; Šimun NALETILIĆ3* (dopisni autor), naletilic@veinst.hr, orcid.org/0009-0002-5805-9892; Barbara IULINI4, barbara.iulini@izsplv.it; Karmen BRANOVIĆ ČAKANIĆ1, branovic@veinst.hr, orcid.org/0000-0001-8760-4680.
1Laboratorij za transmisivne spongiformne encefalopatije, Odjel za patološku morfologiju, Hrvatski veterinarski institut, 10000 Zagreb, Hrvatska
2Laboratorij za bolesti akvatičnih životinja, Odjel za patološku morfologiju, Hrvatski veterinarski institut, 10000 Zagreb, Hrvatska
3Laboratorij za patologiju, Odjel za patološku morfologiju, Hrvatski veterinarski institut, 10000 Zagreb, Hrvatska
4Veterinary Medical Research Institute for Piedmont, Liguria and the Aosta Valley, Laboratory of Neuropathology, 10154 Turin, ItalyNormalni stanični prionski protein (PrPC) prelaskom u pogrešno savijenu patogenu izoformu postaje infektivni prionski protein (PrPSc) koji prouzroči neurodegenerativne bolesti. Prionske bolesti predstavljaju skupinu transmisivnih spongiformnih encefalopatija (TSE), koje mogu nastati uslijed genetskih mutacija, infektivnog prijenosa ili sporadično. Karakterizira ih nakupljanje patološkog prionskog proteina i posljedična neurodegeneracija moždanog tkiva.
U ljudi, prionske bolesti uključuju Kuru, Creutzfeldt-Jakobovu bolest (CJD), varijantu Creutzfeldt-Jakobove bolesti (vCJD), Gerstmann-Sträussler-Scheinkerov sindrom i Fatalnu obiteljsku nesanicu, a u životinja atipičnu i klasičnu goveđu spongiformnu encefalopatiju (GSE), poznatu i kao “kravlje ludilo”, atipični i klasični grebež u ovaca i koza te spongiformnu encefalopatiju jelena. Iako mehanizmi nastanka neurodegenerativnih promjena, kao i uloga upalne reakcije još nisu u potpunosti razjašnjeni, utvrđene su karakteristične patohistološke promjene koje uključuju spongiformne lezije, vakuolizaciju neurona, gliozu i nakupljanje PrPSc proteina. Do danas ne postoji pouzdan zaživotni dijagnostički test za TSE, već se bolest potvrđuje isključivo nakon smrti. Imunohistokemijska pretraga predstavlja jednu od ključnih metoda u dijagnostici TSE-a. Ovaj pregledni članak rezimira dostupne podatke iz literature o prionskim bolestima, s naglaskom na identifikaciju i distribuciju PrPSc u moždanom tkivu goveda, ovaca i koza primjenom imunohistokemijske dijagnostičke metode.
Ključne riječi: prion, TSE, grebež, GS