
Metallothionein gene methylation reflects epigenetic changes in white stork (Ciconia ciconia L., 1758) nestlings
Kalinić, B. Jarić, D. Bjedov, V. Beneš, L. Jurinović, A. Mikuška*
Lidija KALINIĆ, PhD, Associate Professor at Department of Biology, Josip Juraj Strossmayer University of Osijek; Bernard JARIĆ, mag. biol., PhD student at Institute Ruđer Bošković, Zagreb, Croatia; Dora BJEDOV, PhD, Assistant Professor at Department of Biology, Josip Juraj Strossmayer University of Osijek, Croatia; Veronika BENEŠ, univ. bacc. biol., Master student at Department of Biology, Faculty of Science, University of Zagreb, Croatia; Luka JURINOVIĆ, PhD, Senior Scientific Research Associate at Poultry Centre, Croatian Veterinary Institute, Zagreb, Croatia; Alma MIKUŠKA*, PhD, Assistant Professor at Department of Biology, Josip Juraj Strossmayer University of Osijek, Croatia, (Corresponding author, e-mail: amikuska@biologija.unios.hr)
![]() https://doi.org/10.46419/cvj.57.1.5
https://doi.org/10.46419/cvj.57.1.5
Abstract
The white stork (Ciconia ciconia), a migratory bird from the Ciconiidae family, thrives in open habitats such as floodplains and wet meadows. In recent years, they have increasingly turned to foraging at landfills. Given that, white storks are apex predators and environmental pollutants accumulate in their bodies through the food web. Such exposure to pollutants can result in environmental stress, leading to epigenetic modifications that can influence gene expression. In this study, the percentage of the methylation index of the regulatory region of the metallothionein gene was analysed using methylation-specific real-time PCR to investigate the potential epigenetic effects of environmental pollution on white storks. The results suggest that both hypermethylation and hypomethylation of the MT gene may be associated with exposure to specific metals, depending on the pollutant profile and the nestling’s detoxification capacity. Variations in the methylation index point to potential epigenetic responses in white stork nestling populations, which could serve as sensitive indicators of environmental stress. These findings represent the first successful application of DNA methylation analysis in the white stork nestlings in Croatia. Further detailed studies are needed to elucidate the causes of these methylation differences and to explore their relationship with metallothionein gene expression and broader biological outcomes. These results provide a foundation for future research aimed at understanding the influence of environmental pollutants on wildlife health through epigenetic pathways, and highlight the potential of epigenetic tools for wildlife biomonitoring and conservation strategies.
Key words: epigenetics, white stork, methylation-specific real-time PCR, metallothioneins
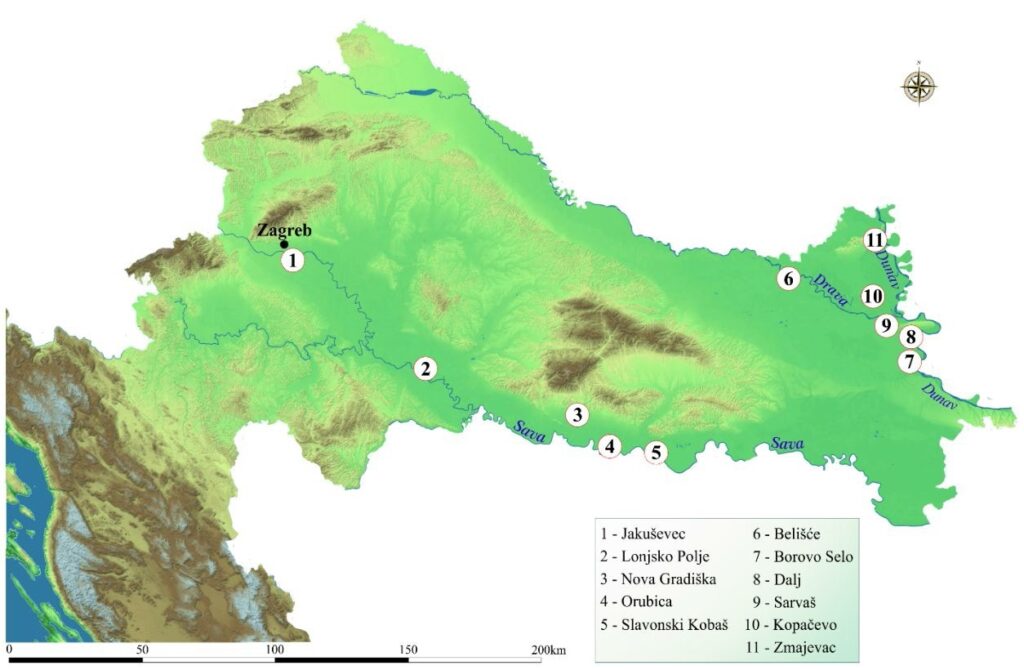
Figure 1. White stork (C. ciconia) nestlings sampling locations in continental Croatia along large rivers: Sava (Jakuševec, Lonjsko Polje, Nova Gradiška, Orubica, Slavonski Kobaš), Drava (Belišće, Sarvaš) and Danube (Borovo Selo, Dalj, Kopačevo, Zmajevac)
DNA isolation and bisulphite conversion
DNA was isolated using 25 µL white stork nestling blood as described in Bjedov et al. (2021). The bisulphite conversion of DNA was performed according to the protocol using the EZ DNA Meth- ylation-Gold Kit (ZYMO Research, Irvine, CA, USA) using 1 µg DNA. The reaction was carried out under the following conditions: 98°C for 8 min, 54°C for 60 min, and a final step at 4°C. Subsequently, 10 µL M-Elution buffer was added, and the samples were centrifuged for 30 s at maximum speed. Following the conversion, the DNA concentration was measured, and the samples were stored at -20°C. The DNA concentration after bisulphite conversion was measured using RNA concentration settings on a nano photometer (Implen, Germany).
Primer synthesis and optimisation
For primer synthesis, MethPrimer (Li and Dahiya, 2002) was used, and four sets each of two specific primers were synthesised: M-primers, which amplify methylated sequences, and U-primers, which amplify unmethylated sequences (Table 1). Primers were synthesised based on the sequence of the regulatory region of the MT gene of the domestic chicken (Gallus gallus domesticus) (Accession number: X13545.1). The primers were designed to amplify methylated and unmethylated regions upstream of the Metal Regulatory Element (Curradi et al., 2002). Optimisation was carried out using Hot Start Taq polymerase. The PCR reaction mixture consisted of 10 µL EmeraldAmp Max HS PCR Master Mix (Takara Bio Europe), 250 nmol forward (FW) primer, 250 nmol reverse (RW) primer, 1 µL bisulphite converted DNA sample (approximately 30 ng DNA), and 8 µL ultrapure wa- ter. For the negative control, nuclease-free water was used in place of the DNA sample. Due to the presence of the Hot Start enzyme, initial denaturation and strand separation at 95°C for 5 to 15 min were not required for the Hot PCR reaction, and a 30 s cycle was sufficient. This was followed by a cycle of 5 s at 98°C, 30 s at the hybridisation temperature (55–62°C), and 1 min at 72°C. After the PCR reaction, PCR products were visualised on 2% agarose gel. The gels were photographed following electrophoresis using Kodak 1D 3.6 soft- ware (Kodak).
Methylation-specific real-time PCR (qMSP)
The methylation index (MI) was determined using qMSP with the EpiScope MSP Kit (Takara).
The reaction mixture consisted of 10 µL MSP buff- er, 250 nmoL each forward (FW) primer, 250 nmoL each reverse (RW) primer, 0.2 µL TB Green solution, 0.48 µL MSP enzyme, 1 µL bisulphite converted DNA sample, and 6.92 µL nuclease-free water. For the negative control, nuclease-free water was used to ensure that there is no contamination with foreign DNA or no cross contamination between samples. The conditions for the qMSP reaction were as follows: initial denaturation at 95°C for 30 s, followed by 40 cycles: denaturation at 98°C for 5 s, primer annealing at 58°C for 30 s, and extension at 72°C for 1 min. Fully methylated (EpiScope® Methylated HCT116 gDNA, Takara Bio Europe) and unmethylated (EpiScope® Unmethylated HCT116 DKO gDNA, Takara) human genomic DNA was used as a standard, amplified with primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (GeneBank NC_000012.12). The primer sequences used were M-primer pair: MF AAATTCGGGAG- GTTAGGGAC, MR CTACAATACGAAAACCGCCC;
U-primer pair: UF AAATTTGGGAGGTTAGGGAT, UR CCCCTACAATACAAAAACCACC
Table 1. Specific primers for amplifying the metallothionein (MT) gene sequence in white stork nestling blood.

Each set includes M-primers for amplifying methylated and U-primers for amplifying unmethylated gene sequences in white stork nestling blood. FW-forward primer; RW-reverse primer; bp-base pairs; Tm-melting temparature
Statistical analysis
All analysis was performed and data was visualised in R 3.6.0 and RStudio 2024.12.0.
Based on the Ct values from the amplification curves, the MI was calculated based on Hattori and Ushijima (2017) using the formula:

where:
M – Ct value for the sample using the M-primer
U – the Ct value for the sample using the U-primer.
Shapiro–Wilks and QQ plots were used to confirm the normality of the data. A one-sample t-test was performed to compare if the sample mean is significantly greater or smaller than the val- ues in the dataset. The following formula was used for calculation:

where:
x̄ – sample mean
μ0 – reference value
s – standard deviation n – sample size.
The Z-score was calculated for each meth- ylation index value to see how distant each value is from the mean in terms of standard deviation.
A positive Z-score indicates a value greater than the mean, and a negative Z-score indicates a value smaller than the mean. The Z-score was calculated based on the formula:

where:
x – individual MI value μ – sample mean
o – sample standard deviation.
Results
Optimisation of primer hybridisation temperature
Following the gradient PCR reaction using four sets of two specific primers, the products were visualised via gel electrophoresis on 2% agarose gel. PCR products were obtained by amplifying sample 127 using primer sets 3 and 4 at temperatures of 60°C, 62°C, and 58°C. Upon gel visualisation, undefined fragments were observed. The gel visualised with sample 127, amplified using specific primers from sets 1 and 2, revealed that both U and M primers exhibited a common hybridisation tem- perature of 58°C, which was found to be optimal.
qMSP – amplification and melting curve analysis
After performing the qMSP reaction, the amplification curve and melting curve were analysed for the M and U primers. The amplification curve indicated that both methylated and unmethylated DNA amplified approximately equally (Fig. 2). The melting curve analysis demonstrated the specificity of the M and U primers at the optimised temperature of 58°C (Fig. 3). As a control for qMSP reaction, human genomic DNA isolated from the HCT116 cell line was used as a standard and amplified using primers for GAPDH. Fig. 2 shows the amplification curves for fully methylated DNA and unmethylated DNA.
Figure 2. Amplification curves (ΔRn vs. Cycle) for human genomic DNA isolated from the HCT 116 cell line (Takara Bio Europe), fully methylated (A) and unmethylated (B).
The melting curve (Tm) for M and U primers used for the amplification of standard DNA (C).

Figure 3. The melting curve (Tm) for M and U primers used for the amplification of white stork DNA samples. Amplification curves were obtained by amplifying white stork DNA samples with M and U primers in the qMSP reaction.

Figure 4. Methylation index (MI, %) of 15 white stork (C. ciconia) samples. The horizontal line at 50% represents the threshold for the reference value, indicating the midpoint between two potential states of methylation or epigenetic marks.
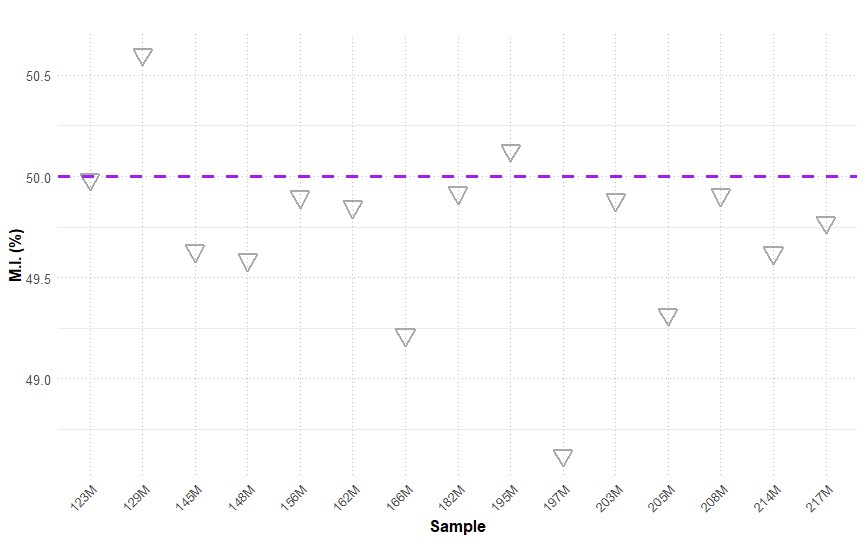
Table 2. Methylation index (MI, %, present study) for each sampling location, nest and sample identification code, with the corresponding metallothionein (MT, mg L-1) plasma concentrations and measured levels of metalloids (arsenic, selenium, µg L-1) and heavy metals (cadmium, mercury, lead, µg L-1) in whole blood of white stork nestlings (Bjedov et al., 2023a).

MI – Methylation index; MT – metallothionein; As – arsenic, Se – selenium, Cd – cadmium; Hg – mercury; Pb – lead
Methylation index
The methylation index was analysed in the blood of 15 white stork nestlings to assess epi- genetic variations associated with environmental exposure. The mean MI was 49.73, with a standard deviation of 0.45 (Fig. 4). Individual sample values are presented in Table 2, alongside corresponding MT plasma concentrations and measured levels of metalloids and heavy metals in whole blood, providing a comprehensive overview of potential associations between epigenetic modifications and pollutant exposure.
A one-sample t-test was conducted to compare the sample mean methylation index against the reference value of 49.73. The t-statistic was 1.69, with a p-value = 0.11, indicating no statistically significant difference between the observed mean and the reference value at the 95% confidence level (Fig. 5).
The Z-score analysis identified one sample with a methylation level significantly different from the mean at the 95% confidence level (Z > 1.96 or Z < -1.96) and a second sample that approached significance. Specifically, sample 197M exhibited a Z-score of -2.47, indicating a significantly lower methylation level compared to the mean (Fig. 6). Additionally, sample 129M had a Z-score of 1.94, suggesting a trend toward significance but not exceeding the threshold for statistical significance (Fig. 6).
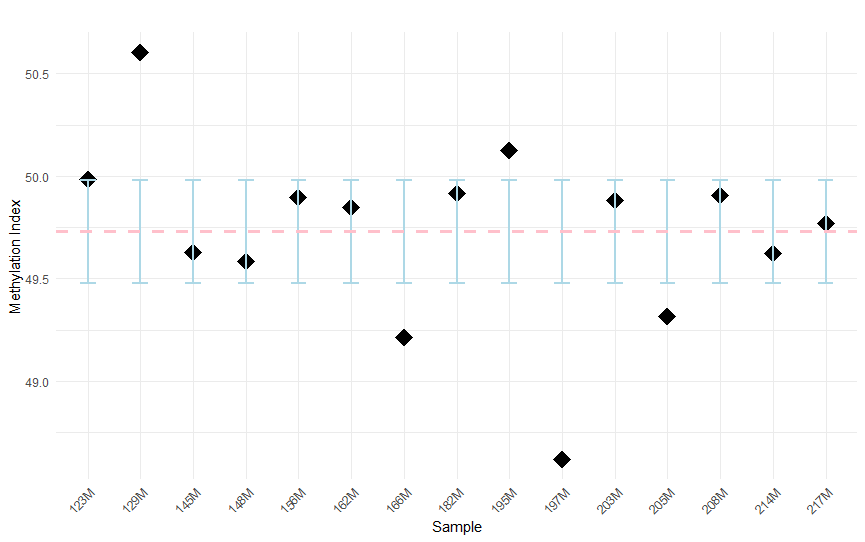
95% CI were calculated for individual methylation indices relative to the reference threshold of 49.73. Samples 129M and 195M exhibited confidence intervals entirely above the threshold, indicating significantly higher methylation levels. In contrast, samples 166M, 197M and 205M had confidence intervals entirely below the threshold, indicating significantly lower methylation. All other samples had confidence intervals that included the reference value, indicating no significant deviation. These results identify five samples with statistically significant differences in methylation relative to the reference value (Fig. 7).
Figure 5. Histogram of methylation index (%) for the white stork (C. ciconia) samples, plotted to assess the distribution relative to the reference value of 49.73 shown by the vertical dashed line, using the one-sample t–test.
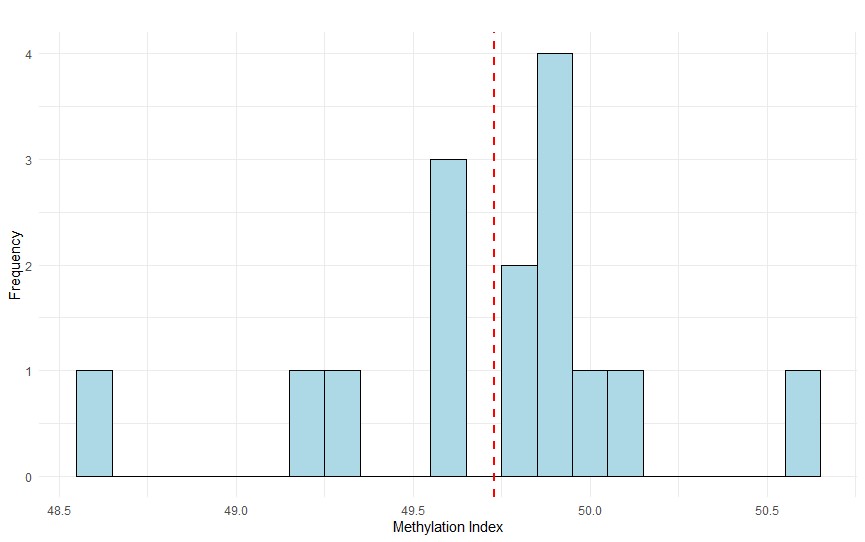
Figure 6. Z-scores of the methylation index for white stork (C. ciconia) samples, calculated to standardise the data relative to the mean and standard deviation of the sample set. The Z-score transformation allows for the comparison of each sample’s methylation index in terms of how many standard deviations it deviates from the overall mean. A Z-score of 0 indicates a sample with a methylation index exactly equal to the mean, while positive or negative Z-scores represent samples above or below the mean, respectively.

Figure 7. Confidence intervals for the methylation indices of white stork (C. ciconia) samples, calculated at a 95% confidence level. Each point represents the mean methylation index for an individual sample, with the vertical error bars indicating the range of values within which the true mean is expected to fall with 95% certainty.
Discussion
Epigenetics prioritises the study of how environmental exposures affect gene regulation throughout the life of organisms. Long-term ex- posure to stressors is known to create lifelong changes in stress sensitivity that are associated with epigenetic changes (Jensen, 2014). Epigenetic profiles in blood cells have been shown to reflect methylation patterns associated with other tissues, such as the liver, making blood a useful non-invasive biomarker (Laine et al., 2021). Also, research has confirmed that stress affects DNA methylation in blood cells (Pértille et al., 2017).
Methylation levels were significantly higher in the blood of young birds compared to embryonic tissue at any stage of prenatal development, provid- ing evidence that DNA undergoes global changes during development in birds and supporting the hypothesis that methylation mediates phenotypic development. Research indicates that the largest amount of modified DNA comes precisely from erythrocytes in the blood (Watson et al., 2019). By collecting samples, isolating and analysing DNA from the blood of white stork nestlings, we gathered information about the level of epigenetic modifications, i.e., DNA methylation of the CpG island of the metallothionein gene. One of the methods that can be used to determine the methylation index of certain genes is qMSP. This method is sensitive and specific for determining methylated DNA sequences (Hattori and Ushijima, 2017).
It can distinguish methylated from unmethylated parts using short oligonucleotide primers whose 3′ ends correspond to specific methylated CpG sites in the sample. Following bisulfite conversion, qMSP employs methylation-specific primers and an intercalating fluorescent dye to quantify amplification. It indicates the number of cycles in which expression occurs (Ct value). The Ct value is inversely proportional to the expression or methylation of CpG islands. This would mean that lower Ct values represent the quantity of methylated parts already at a small number of cycles (Husseiny et al., 2012).
The present study determined the MI of the regulatory region of the gene for MT in the blood of the white stork nestlings. MTs contain a palindro- mic sequence Metal Responsive Element (MRE) in their promoter region near the TATA box (cis-acting region), which is activated by the binding of the MTF-1 protein. Metals in the environment and exposure to oxidative stress promote the binding of MTF-1 to DNA, after which the genes respon- sible for MT protein synthesis are activated. MTs are responsible for protecting the body from toxic metals and oxidative stress. The analysis revealed that five samples exhibited statistically significant differences in DNA methylation levels (Table 2, Fig. 7), where samples 129M and 195M showed significantly elevated MI values, while samples 166M, 197M, and 205M displayed significantly reduced MI values. The elevated MI in samples 129M and 195M may be associated with exposure to As, Se, and Hg, which were elevated in these samples (Table 2) and to Se (1470 µg L-1), while 195M showed elevated Hg (34.80 µg L-1). These findings suggest that As may promote hypermethylation through disruption of one-carbon metabolism or DNMT activity. In contrast, significantly lower methylation in samples 166M, 197M, and 205M may reflect hypomethylation associated with genotoxic stress or impaired epigenetic regulation in response to metal(loid) exposure. Notably, these samples exhibited some of the highest concentrations of Hg (90.40 µg L-1 in 197M), Pb (67.20 µg L-1 in 205M), and As (74.70 µg L-1 in 166M), along with relatively low MT levels (Table 2). Given the MT role in metal detoxification and oxidative stress defence, reduced MT methylation may contribute to insufficient cellular protection, leading to DNA hypomethylation. These patterns suggest that both hyper- and hypomethylation may occur in re- sponse to metal exposure, depending on the spe- cific pollutant profile and the white stork nestling’s capacity for detoxification.
Environmental pollution has a similar effect on the MT genes as on the genes encoding DNMT. It has been shown that an increased concentration of metals in the environment can lead to increased expression of the gene for DNMT (DNMT3), which can lead to de novo methylation (Lyko, 2018).
Stressors such as metal exposure (such as Cu) and temperature, separately or in combination, significantly enhanced the expression of the de novo gene for DNMT (Dorts et al., 2016). The anal- ysis revealed a negative correlation between gene expression and DNA methylation, and increasing methylation decreases the expression of genes responsible for liver cells. This is supported by research that indicates a connection between modifications in the liver and blood cells (Laineet al., 2021). One of the mechanisms of gene expression silencing is through the MeCP2 protein (methyl-CpG-binding protein 2), which binds methylated DNA and inhibits transcription, and research has indicated that various disorders in birds are the result of a mutation of this protein (Kriaucionis and Bird, 2003). Laine et al. (2021) indicated the impact of As on epigenetic changes. Furthermore, during the biotransformation of As, reactive oxygen species (ROS) are generated, which affect epigenetics by creating aberrant methylations.
For the white stork, choosing an appropriate nesting site is crucial for successful reproduction. Due to anthropogenic influences, there are fewer and fewer natural areas for high-quality foraging,and storks are often forced to use artificial for- aging sites, like landfills. Recently, white storks from Western Europe have been using landfills as a place to feed and nest (López-García et al., 2021; Bjedov et al., 2022, 2023a). This behaviour, caused by human influence, carries with it numerous stress factors. As mentioned, stress in early development can affect stress responses later in life. Kang et al. (2017) proposed a mechanism of DNA methylation involved in acute and chronic stress responses of birds. During stress, DNMTs (DNMT3a and DNMT3b) causes causes de novo DNA methylation. When DNA replication occurs under stress, DNA methylation is maintained by DNMT1, which prefers hemi-methylated DNA.
MTs play a vital role in metal homeostasis, detoxification, and cellular defence against oxidative stress. Functioning primarily as metal chelators, MTs bind and regulate essential and non-essential metal ions, including Zn, Cu, and Cd, thereby facilitating their transport, storage, and detoxification (Babula et al., 2012). Their expression is highly inducible by metal exposure, with silver (Ag), Cu, and Cd being the most potent inducers, while other metals such as Hg and Pb may also modulate MT levels in a species and tissue-specific manner (Babula et al., 2012). MT expression is particularly pronounced in organs involved in metal sequestration, including the liver, kidneys, and blood, with Cd among the strongest inducers (Scheuhammer and Templeton, 1990).
However, the degree of MT inducibility varies across tissues, reflecting differential metal accumulation and physiological demands (Elliott et al., 1992). In white stork nestlings, MTs were analysed as potential biomarkers of environmental pollution exposure (Bjedov et al., 2023a). Despite exposure to varying pollutant sources across different sampling localities, no statistically significant differences in MT levels were observed (Bjedov et al., 2023a). This suggests that MT expression in white stork nestlings may be less responsive to environmental pollutants compared to other biomarkers. However, MTs have been extensively studied in other avian species, particularly terrestrial birds, where they have been quantified in the kidney, liver and blood, of white-tailed eagle Haliaeetus albicilla (Marcinekova et al., 2019), ringed turtle dove Streptopelia risoria (Scheuhammer and Tem- pleton, 1990), and great tit Parus major (Vanparys et al., 2008). Similarly, research on aquatic bird species has demonstrated the presence of MTs in the double-crested cormorant Phalacrocorax auritus, Atlantic puffin Fratercula arctica, Leach’s storm-petrel Oceanodroma leucorhoa, and herring gull Larus argentatus (Elliott et al., 1992). Addition- al studies have documented MT levels in the great cormorant Phalacrocorax carbo, Indian spot-billed duck Anas poecilorhyncha and mallard Anas platy- rhynchos (Nam et al., 2005). Due to their role in metal detoxification, MTs are frequently analysed alongside key metal pollutants such as Cd, Cu and Zn, facilitating ecotoxicological assessments of pollution and physiological responses. This approach has been effectively employed in the white-winged scoter Melanitta fusca and surf scoter Melanitta perspicillata, where MT levels were measured in parallel with these metals to assess environmental exposure and potential physiological impacts (Barjaktarovic et al., 2002). Future research could benefit from similar methodologies, providing deeper insights into (heavy) metal exposure and its physiological consequences in white stork adult and nestling individuals.
Conclusion
The present study shows that both hyper- methylation and hypomethylation in white stork (C. ciconia) nestlings may be associated with exposure to specific metals, depending on the pollutant profile and the nestling’s detoxification capacity. Variations in the DNA methylation index observed suggest that epigenetic responses in wild bird populations may serve as sensitive indicators of environmental stress. Importantly, this study represents the first successful application of DNA methylation analysis in a wild bird population in Croatia, and the first time such an approach has been used on white stork nestlings in this region. Given the complexity of epigenetic mechanisms and their potential ecological relevance, further detailed study is needed to determine the exact causes of observed methylation differences, and to assess their relationship with metallothionein gene expression and broader biological outcomes. The present findings should be viewed as preliminary, serving as a foundation for future research aimed at understanding how environmental pollutants influence the health and development of wildlife through epigenetic pathways, and exploring the potential of epigenetic tools in wildlife biomonitor- ing and conservation strategies.
Acknowledgements
This study was funded by the Department of Biology, Josip Juraj Strossmayer University of Osijek, research project: Research of native and non-native fauna of birds, fish and insects, and bi- oaccumulation of heavy metals in their food webs (3105-4-23). We are thankful to the electricity provider HEP from Osijek and Zagreb who provided us with the telescopic crane and manpower to access stork nests, and to Tibor Mikuska B.Sc. as the offi- cial bird ringer from the Croatian Ringing Centre at the Croatian Academy of Science and Arts, Zagreb.
References [… show]
Metilacija gena za metalotionein odražava epigenetske promjene kod gniježđenja bijele rode (Ciconia ciconia L., 1758)
Dr. sc. Lidija KALINIĆ, izvanredna profesorica, Odjel za biologiju, Sveučilište Josipa Jurja Strossmayera u Osijeku, Osijek, Hrvatska; Bernard JARIĆ, mag. biol., magistar biologije, Institut Ruđer Bošković, Zagreb, Hrvatska; dr. sc. Dora BJEDOV, docentica, Odjel za biologiju, Sveučilište Josipa Jurja
Strossmayera u Osijeku, Osijek, Hrvatska; Veronika BENEŠ, univ. bacc. biol., prvostupnica biologije, Odjel za biologiju, Sveučilište Josipa Jurja Strossmayera u Osijeku, Osijek, Hrvatska; dr. sc. Luka JURINOVIĆ, viši znanstveni suradnik, Centar za peradarstvo, Hrvatski veterinarski institut, Zagreb, Croatia; dr. sc.
Alma MIKUŠKA, docentica, Odjel za biologiju, Sveučilište Josipa Jurja Strossmayera u Osijeku, Osijek, Hrvatska.
Bijela roda (Ciconia ciconia L., 1758), migratorna ptica iz porodice Ciconiidae, nastanjuje otvorena i vlažna staništa poput poplavnih nizina i vlažnih livada. U posljednjim godinama, sve više se hrani na odlagalištima otpada. S obzirom na to kako su bijele rode vršni i predatori, okolišna zagađivala akumuliraju se putem hranidbenih mreža u njihovim tijelima. Izloženost zagađivalima može izazvati fiziološki stres, što dovodi do epigenetskih promjena koje mogu utjecati na ekspresiju gena. U ovom istraživanju analiziran je postotak indeksa metilacije (M. I., %) regulatorne regije gena za metalotionein (MT) u krvi ptića bijele rode kako bi se ispitali potencijalni epigenetski učinci okolišnog zagađenja korištenjem metilacijom specifične real-time PCR (qMSP) tehnike. Rezultati sugeriraju da hipermetilacija i hipometilacija gena MT mogu biti povezane s izloženošću specifičnim metalima, ovisno o profilu zagađivala i sposobnosti ptića za detoksifikaciju. Varijacije u M.I. ukazuju na potencijalne epigenetske odgovore u populacijama ptića bijele rode, koji bi mogli poslužiti kao osjetljivi indikatori okolišnih stresora. Ovi rezultati predstavljaju prvi uspješan primijenjeni pristup metilaciji DNA na ptićima bijele rode u Hrvatskoj. Daljnja istraživanja nužna su kako bi se razjasnili uzroci razlika u metilaciji i ispitala njihova povezanost s ekspresijom gena za MT i širim biološkim posljedicama. Ova istraživanja pružaju temelj za daljnja istraživanja koja će omogućiti bolje razumijevanje utjecaja okolišnih zagađivala na zdravlje divljih životinja putem epigenetskih mehanizama te ističu potencijal epigenetskih alata u biomonitoringu i strategijama očuvanja divljih vrsta.
Ključne riječi: epigenetika, bijela roda, meti- lacijsko-specifični real-time PCR, metalotioneini